M.I. Gobes, Y. Buğday, J.P.M. van Duynhoven, J. Hohlbein, R.G.M. van der Sman, Food Hydrocolloids, 174, 112316, 2026, [link]
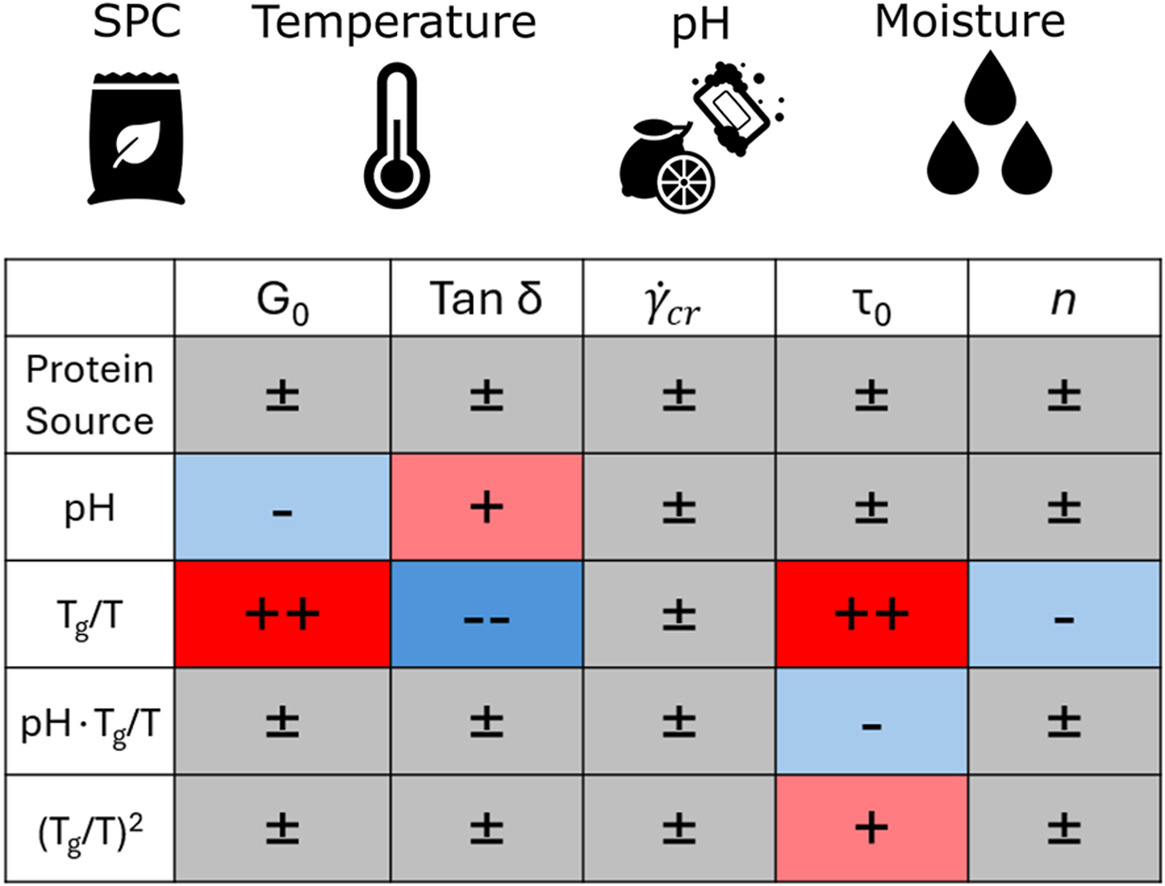
The texture of plant-based-meat analogues manufactured by high-moisture extrusion is governed by the rheological properties of the extruded protein dough. However, there is currently little understanding of how dough formulation and temperature impact these properties. Characterisation of rheology during steady-shear under extrusion-relevant conditions is challenging; hence, we performed large amplitude oscillatory-shear (LAOS) rheology using a closed-cavity rheometer to reach relevant temperatures and pressures, and characterised soy protein concentrate doughs while varying protein source, pH, moisture content, and temperature. Steady-shear behaviour was extracted from LAOS using the sequence of physical processes. We then fitted a constitutive Herschel-Bulkley model to the extracted steady-shear data and a descriptive model to the strain-sweep data. Using multiple linear regression, we link the fitted parameters to the experimental conditions. The regression shows that the temperature and moisture effects can be aggregated via the ratio Tg/T, with Tg being the moisture-dependent glass-transition temperature, and T the actual temperature. The yield stress τ0, elastic modulus G0, and the loss factor tan(δ) were influenced significantly by Tg/T and, to a lesser degree, by the pH. Parameters describing rheology at large deformations, the critical shear rate and the strain-thinning index n, are not affected by any experimental condition. Despite significant differences in protein solubility between the two analysed concentrates, we observed no significant difference between the concentrates after shifting them to the same pH. Our work quantifies the dependencies of the parameters behind the rheological behaviour of doughs and will enable flow modelling of these doughs in extruder cooling dies.