V. Khandan, V. Boerkamp, A. Jabermoradi, M. Fontana, J. Hohlbein, E. Verpoorte, R.C. Chiechi, K. Mathwig, 2022, pre-print on arXiv, [link]
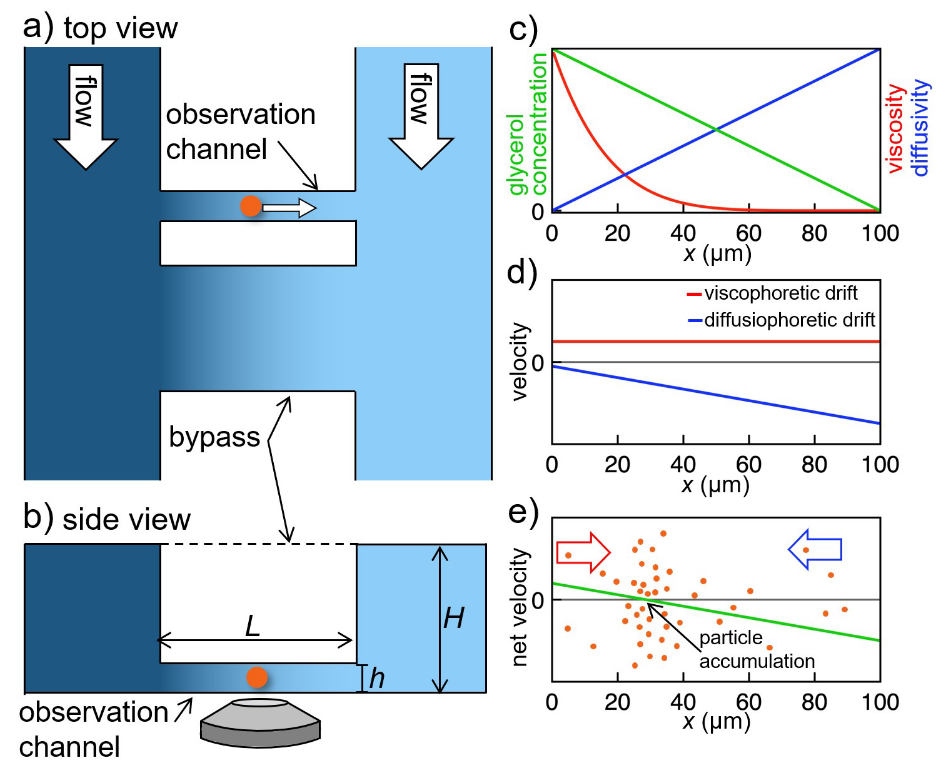
Viscosity is a fundamental property of liquids. It determines transport and diffusion of particles in solution. Nonetheless, it is an open question how a gradient of viscosity – causing a gradient in diffusivity – can lead to viscophoretic transport, i.e., directed transport of particles and molecules in solution. Here, we determine viscophoretic drift experimentally. We generate steep, stable viscosity gradients in a microfluidic device and image transport of suspended nanoparticles in these gradients using high-resolution microscopy. We observe high viscophoretic drift velocities which significantly exceed theoretical predictions. In addition, we demonstrate a new method for trapping and concentrating particles by using the interplay of viscophoresis and diffusiophoresis. We believe that a quantification of viscophoresis will advance the understanding and application of transport processes of gradients of viscosity occurring in very diverse fields such as cell biology, chromatography, and membrane technology.