This website is for legacy purposes only and won’t be updated much longer. You can find us now at our new website at the BTU Cottbus-Senftenberg: https://www.b-tu.de/en/fg-biosensorik/page
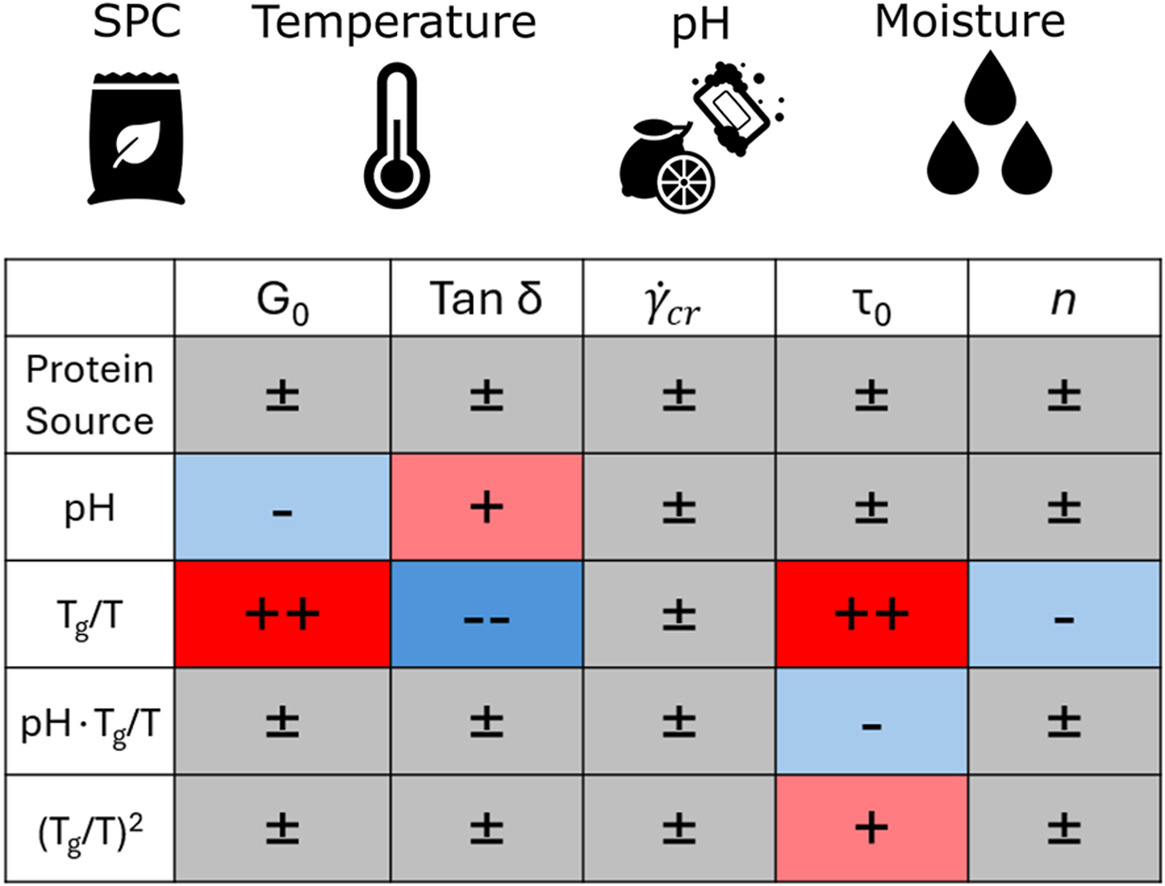
Published: Impact of pH, temperature, and moisture on the rheological behaviour of soy protein concentrate doughs
M.I. Gobes, Y. Buğday, J.P.M. van Duynhoven, J. Hohlbein, R.G.M. van der Sman, Food Hydrocolloids, 174, 112316, 2026, [link]
The texture of plant-based-meat analogues manufactured by high-moisture extrusion is governed by the rheological properties of the extruded protein dough. However, there is currently little understanding of how dough formulation and temperature impact these properties. Characterisation of rheology during steady-shear under extrusion-relevant conditions is challenging; hence, we performed large amplitude oscillatory-shear (LAOS) rheology using a closed-cavity rheometer to reach relevant temperatures and pressures, and characterised soy protein concentrate doughs while varying protein source, pH, moisture content, and temperature. Steady-shear behaviour was extracted from LAOS using the sequence of physical processes. We then fitted a constitutive Herschel-Bulkley model to the extracted steady-shear data and a descriptive model to the strain-sweep data. Using multiple linear regression, we link the fitted parameters to the experimental conditions. The regression shows that the temperature and moisture effects can be aggregated via the ratio Tg/T, with Tg being the moisture-dependent glass-transition temperature, and T the actual temperature. The yield stress τ0, elastic modulus G0, and the loss factor tan(δ) were influenced significantly by Tg/T and, to a lesser degree, by the pH. Parameters describing rheology at large deformations, the critical shear rate and the strain-thinning index n, are not affected by any experimental condition. Despite significant differences in protein solubility between the two analysed concentrates, we observed no significant difference between the concentrates after shifting them to the same pH. Our work quantifies the dependencies of the parameters behind the rheological behaviour of doughs and will enable flow modelling of these doughs in extruder cooling dies.
Published: Mapping anisotropic structure formation of soy protein during high-moisture extrusion
E.D. Garina, S.A. Kuijpers, M.I. Gobes, A. Sein, R. den Adel, G.N. Smith, M. Sztucki, J. Hohlbein, C. Terenzi, J.P.M. van Duynhoven, W.G. Bouwman, Food Hydrocolloids, 172, 112037, 2026, [link]
The development of novel plant-based meat alternatives that closely mimic the anisotropic structure of animal meat offers a solution to mitigate the adverse effects of animal meat consumption. The currently most widely adopted production route is shear processing through high-moisture extrusion (HME). The complex structure formation mechanisms that determine the final fibrous texture of extrudates have yet to be fully understood. The main obstacle is the lack of multiscale studies investigating the principles governing structure formation from the nano- to the macro-structural level. This work aims to address this knowledge gap by studying materials, collected after a dead-stop operation of an industrial pilot-plant scale extruder, with multiple characterisation techniques, such as Magnetic Resonance Imaging (MRI) and Small-Angle Scattering (SAS). We demonstrate that the nm- to µm-scale structure is formed already within the extruder barrels, and that sub-mm-scale anisotropy develops within the cooling die. Furthermore, we show that diffuse light reflectance (DR) probes the size and coarseness of the lamellar phase-separated regions.

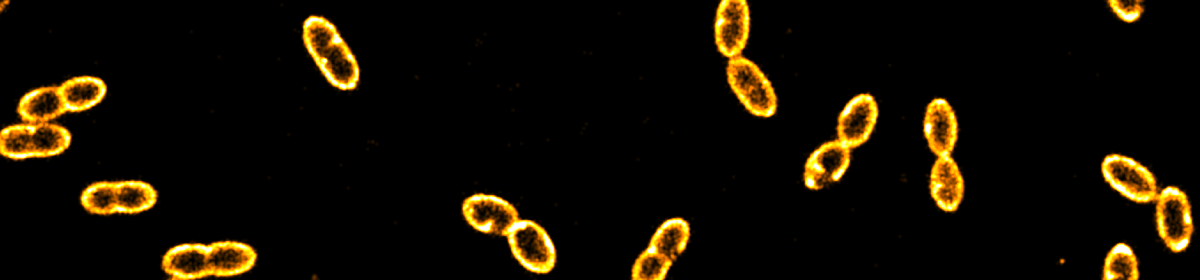
Published: The impact of DnaA titration on Escherichia coli DNA replication initiation elucidated through live-cell single-particle tracking
L. Olivi, S. Köstlbacher, C. Ludwig, M. Langendoen, N.J. Claassens, T.J.G. Ettema, , J. van der Oost, P.R. ten Wolde, J. Hohlbein*, R. Staals*, Nature Communications, 16, 7813, 2025, [link], preprint on bioRxiv, [link]
DNA replication initiation is orchestrated in bacteria by the replication initiator DnaA. Two models for regulation of DnaA activity in Escherichia coli have been proposed: the switch between an active and inactive form, and the titration of DnaA on the chromosome. Although proposed decades ago, experimental evidence of a titration-based control mechanism is still lacking. Here, we first identified a conserved high-density region of binding motifs near the origin of replication, an advantageous trait for titration of DnaA. We then investigated the mobility of DnaA by visualising single proteins inside single cells of wild-type and deletion mutants E. coli strains, while monitoring cellular size and DNA content. Our results indicate that the chromosome of E. coli controls the free amount of DnaA in a growth rate-dependent fashion. Moreover, they address long-standing questions on the relevance of DnaA titration in stabilising DNA replication by preventing re-initiation events during slow growth.

Published: Rotated Fourier transform (RFT) enables the quantification of anisotropic structure in high-moisture plant-protein extrudates
M.I. Gobes, S.A. Kuijpers, C. Terenzi, R.G.M. van der Smaan, J.P.M. van Duynhoven, J. Hohlbein, Food Structure, 44, 100437, 2025, [link], preprint on chemRxiv, [link]
When producing plant-protein-based meat analogues via high moisture extrusion (HME), the structure of extrudates is determined by complex interactions between ingredient composition and processing conditions. Insights into the structuring process can be gained by imaging samples using MRI or confocal microscopy. However, existing software for analysing these images provide limited options for quantitatively analysing both structure and anisotropy. Here, we present a novel image processing method, Rotated Fourier Transform (RFT), that enables the quantification of anisotropic structures of extrudates. RFT provides a single, spatial dependent measure of structural anisotropy, namely the weighted order parameter (WOP). RFT uses Fourier transforms to obtain the dominant angles representing the structural orientation detected within the image. By calculating an amplitude per angle as the weighing factor, noise is effectively filtered and improved signal-to-noise ratios can be obtained. In particular, we applied RFT to quantify structural anisotropy of soy protein concentrate HME samples. We employed magnetic resonance imaging (MRI) at micrometer resolution to show that samples prepared at neutral pH feature higher structural anisotropy than samples prepared at acidic pH. Using confocal laser scanning microscopy (CLSM) at sub-micrometer resolution, we imaged samples from the skin to the core region along the cooling die and show that the anisotropy increases towards the skin. We note that RFT is a generic method applicable to any image displaying anisotropic features. Thus, RFT is a powerful tool for the comprehensive quantification of food structures and beyond.

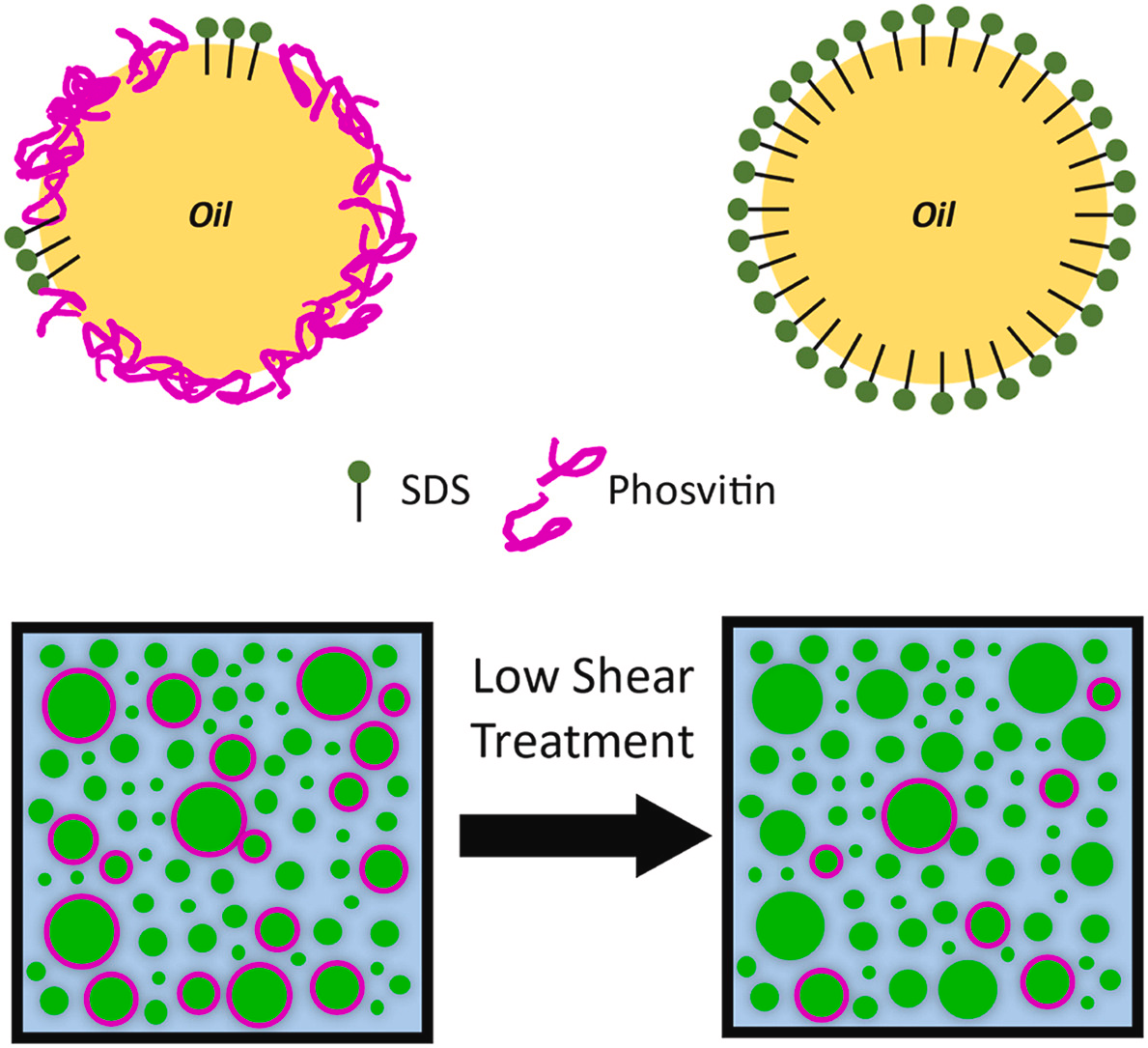
Published: Super-resolution imaging reveals heterogeneity in the coverage of oil-in-water food emulsions
A. Jabermoradi, S. Foroutanparsa, I.K. Voets, J.J.M. Janssen, J.P.M. van Duynhoven, and J. Hohlbein, Food Hydrocolloids, 168, 111490, 2025, [link]
Oil-in-water food emulsions such as mayonnaise and dressings are stabilized by proteins and low-molecular weight surfactants binding to the oil/water interface. One common source of emulsifying proteins is egg yolk containing the iron-binding protein phosvitin. Here, we applied super-resolution microscopy to quantify the distribution of phosvitin on the droplet interfaces of binary SDS/phosvitin model emulsions prepared by high-pressure homogenization (HPH). We targeted phosvitin either via fluorescently labeled, primary antibodies or with affimers, which are short polypeptides. Re-scan confocal microscopy (RCM) revealed a bimodal droplet size distribution in which small droplets were primarily covered by SDS and large droplets by phosvitin. This inter-droplet heterogeneity was in line with expected kinetics of emulsifier coverage of droplet interfaces during HPH. Stochastic optical reconstruction microscopy (STORM) indicated that changing the concentration of phosvitin did not affect the intra-droplet distribution at the droplet interface. STORM further provided a direct visualization of the redistribution of phosvitin upon prolonged low shear treatment, resulting in diffusion-assisted exchange of SDS and phosvitin between droplet interfaces and the continuous aqueous phase. Our RCM- and STORM-based approaches allow a direct and quantitative view on the intricate balance between kinetic and thermodynamic forces governing inter- and intra-droplet interfacial distributions of proteins.
Published: Impact of pH-shifting on multiscale structural anisotropy of high-moisture extrudates of soy proteins
S.A. Kuijpers, E.D. Garina, M.I. Gobes, R. den Adel, G.N. Smith, M. Sztucki, J. Hohlbein, W.G. Bouwman, J.P.M. van Duynhoven, C. Terenzi, Food Hydrocolloids, 168, 111456, 2025, [link]
High-moisture extrusion (HME) is a proven industrial food processing technique used to create textured plant-protein materials that can serve as alternatives for animal meat. The required multiscale anisotropic structure of the extrudate can be achieved by selecting suitable HME process conditions, as well as by pH-shifting. In this work, we explored pH-shifting via the water feed, which is an attractive industrially-scalable approach. Soy protein concentrate (SPC) was extruded on lab-scale and extrudates were characterized ex situ, from molecular to mm scale, using Diffuse Reflectance (DR), Magnetic Resonance Imaging (MRI), Small-Angle-Scattering of Neutrons (SANS) or X-rays (SAXS). pH-shifting had a non-monotonic effect on extrudate hardness and anisotropic structure at both sub-mm (MRI) and μm (DR) scale. At the sub-μm scale, SANS and SAXS data indicated that, at pH > pI, the radius of protein nano-aggregates monotonically increases, accompanied by a transition from particulate to fibrillar protein aggregation. When pH was further shifted to alkaline conditions, the decrease in clustering strength and nematic order parameter pointed to an increase in intra- and inter-fibrillar repulsion, respectively. Protein extractability experiments indicated that the effects of pH-shifting on anisotropic structure formation could not be attributed to covalent intermolecular crosslinking. Thus, repulsive non-covalent electrostatic protein-protein interactions play a dominant role in the formation of multiscale anisotropic structure during SPC extrusion. The formation of an optimal anisotropic SPC extrudate structure is determined by the pH-dependent balance between fibrillar nano-aggregate clustering and electrostatic repulsion. Alkalization or acidification via the water feed implies that protein charge and structure may not be in equilibrium yet with the imposed pH conditions. The transient nature of pH-shifting via the water feed results in an intricate interplay with extrusion conditions. Therefore, control of anisotropic structure formation, via the water feed, in SPC extrudates, is extruder specific.

Published: Quantifying the distribution of proteins at the interface of oil-in-water food emulsions
Emulsifiers play an essential role in ensuring the physiochemical stability of food emulsions. In the case of mayonnaise, proteins contained in egg yolk act as emulsifiers. Here, we employed stochastic optical reconstruction microscopy (STORM) to localize proteins at the oil/water droplet interface using fluorescently labeled antibodies. To quantitatively analyze the distribution of proteins, we first simulated homogeneous and heterogeneous distributions. We then implemented the relative position distribution (RPD) analysis to extract the histogram of relative distances between all neighboring localizations. By analyzing the local maxima of the histogram, we could classify distributions at droplet interfaces as homogeneous, partially heterogeneous, and heterogeneous. The model fitting over the RPD histogram using a 2D probability function further provided a localization precision amplitude consistent with the analysis of the local maxima. As a model system for mayonnaise, we used emulsions prepared with combinations of phosvitin, phospholipids, apolipoprotein B (apoB), and sodium dodecyl sulfate (SDS) as emulsifiers. The binary phosvitin/SDS model emulsion showed a partially heterogeneous distribution of phosvitin around the droplets. The ternary phosvitin/phospholipid/SDS and apoB/phospholipid/SDS emulsions showed increased heterogeneity of phosvitin and apoB. Quantification of heterogeneity at droplet interfaces may provide insights in factors determining the physical and chemical stability of emulsions.

Published: Spatiotemporal assessment of protein and lipid oxidation in concentrated oil-in-water emulsions stabilized with legume protein isolates
M. Brüls-Gill, V. Boerkamp, J. Hohlbein*, J.P.M. van Duynhoven*, Current Research in Food Science, 9, 100817, 2024, [link],
The growing trend of substituting animal-based proteins with plant-based proteins requires more understanding of the functionality and stability of vegan mayonnaises, especially regarding their susceptibility to lipid and protein oxidation. Here, we investigate the spatial and temporal dynamics of lipid and protein oxidation in emulsions stabilized with legume ((hydrolyzed) soy, pea, and faba bean) protein isolates (hSPI, SPI, PPI, FPI). We assessed lipid oxidation globally by NMR and locally by confocal laser scanning microscopy using the oxidation-sensitive fluorescent dye BODIPY 665/676. Further, we assessed local protein oxidation by employing protein autofluorescence and the fluorescently labelled radical spin-trap CAMPO-AFDye 647. Oxidation of oil in droplets was governed by the presence of tocopherols in the oil phase and pro-oxidant transition metals that were introduced via the protein isolates. Non-stripped oil emulsions stabilized with PPI and hSPI displayed higher levels of lipid hydroperoxides as compared to emulsions prepared with SPI and FPI. We attribute this finding to higher availability of catalytically active transition metals in PPI and hSPI. For stripped oil emulsions stabilized with SPI and FPI, lipid hydroperoxide concentrations were negligible in the presence of ascorbic acid, indicating that this agent acted as antioxidant. For the emulsions prepared with PPI and hSPI, lipid hydroperoxide formation was only partly inhibited by ascorbic acid, indicating a role as prooxidant. Interestingly, we observed protein-lipid aggregates in all emulsions. The aggregates underwent fast and extensive co-oxidation which was also modulated by transition metals and tocopherols originating from the oil phase. Our study demonstrates the potential of spatiotemporal imaging techniques to enhance our understanding of the oxidation processes in emulsions stabilized with plant proteins.

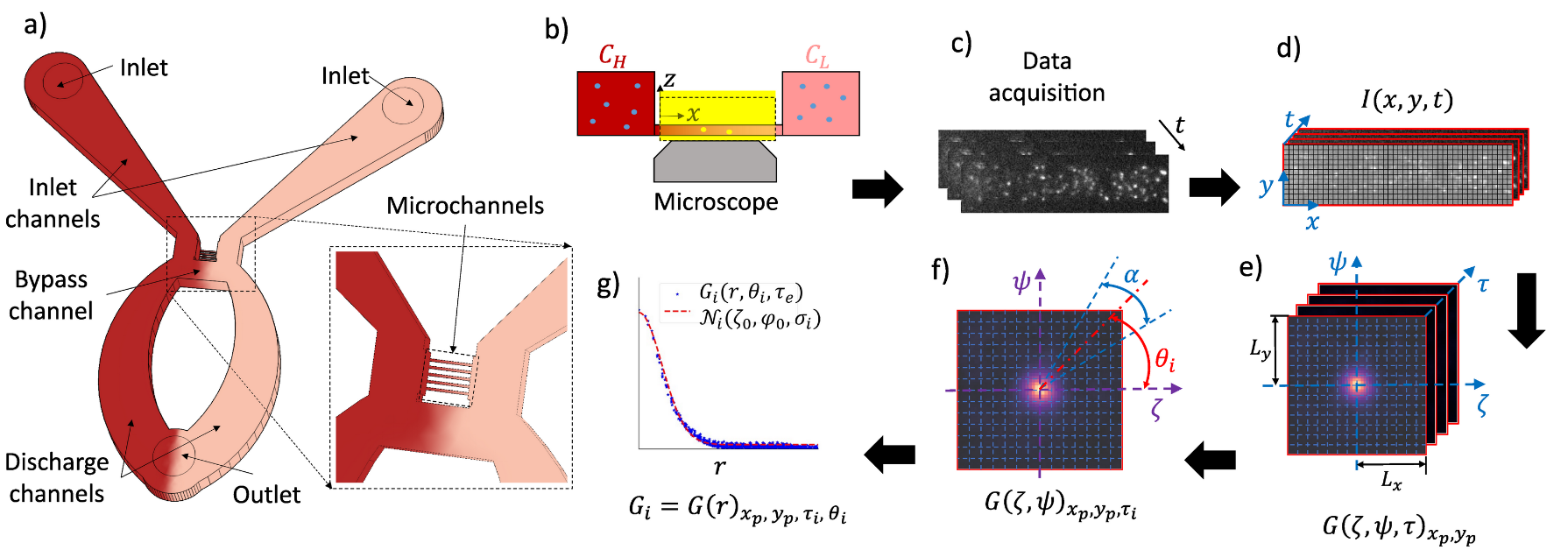
Published: Addressing spatiotemporal signal variations in pair correlation function analysis
V. Khandan, V.J.P. Boerkamp, R.C. Chiechi, J. Hohlbein*, and K. Mathwig*, Biophysical Journal, 2024, [link]
Fluorescence Correlation Spectroscopy (FCS) is a cornerstone technique in optical microscopy to measure, for example, the concentration and diffusivity of fluorescent emitters and biomolecules in solution. The application of FCS to complex biological systems, however, is fraught with inherent intricacies that impair the interpretation of correlation patterns. Critical among these intricacies are temporal variations beyond diffusion in the quantity, intensity, and spatial distribution of fluorescent emitters. These variations introduce distortions into correlated intensity data, thus compromising the accuracy and reproducibility of the analysis. This issue is accentuated in imaging-based approaches such a Pair Correlation Function (pCF) analysis due to their broader Regions of Interest (ROIs) compared to point-detector-based approaches. Despite ongoing developments in FCS, attention to systems characterized by a spatiotemporal-dependent probability distribution function (ST-PDF) has been lacking. To address this knowledge gap, we developed a new analytical framework for ST-PDF systems that introduces a dual-timescale model function within the conventional pCF analysis. Our approach selectively differentiates the signals associated with rapid processes, such as particle diffusion, from signals stemming from spatiotemporal variations in the distribution of fluorescent emitters occurring at extended delay timescales. To corroborate our approach, we conducted proof-of-concept experiments on an ST-PDF system, wherein the, initially, uniform distribution of fluorescent microspheres within a microfluidic channel changes into a localized accumulation of microspheres over time. Our framework is offering a comprehensive solution for investigating various phenomena such as biomolecular binding, sedimentation, and particle accumulation.