S. Yang, S. ten Klooster, K. Nguyen, M. Hennebelle, C. Berton-Carabin, K. Schroën, J. van Duynhoven, J. Hohlbein, Food Research International, 188, 114341, 2024, [link], preprint on chemRxiv, [link]
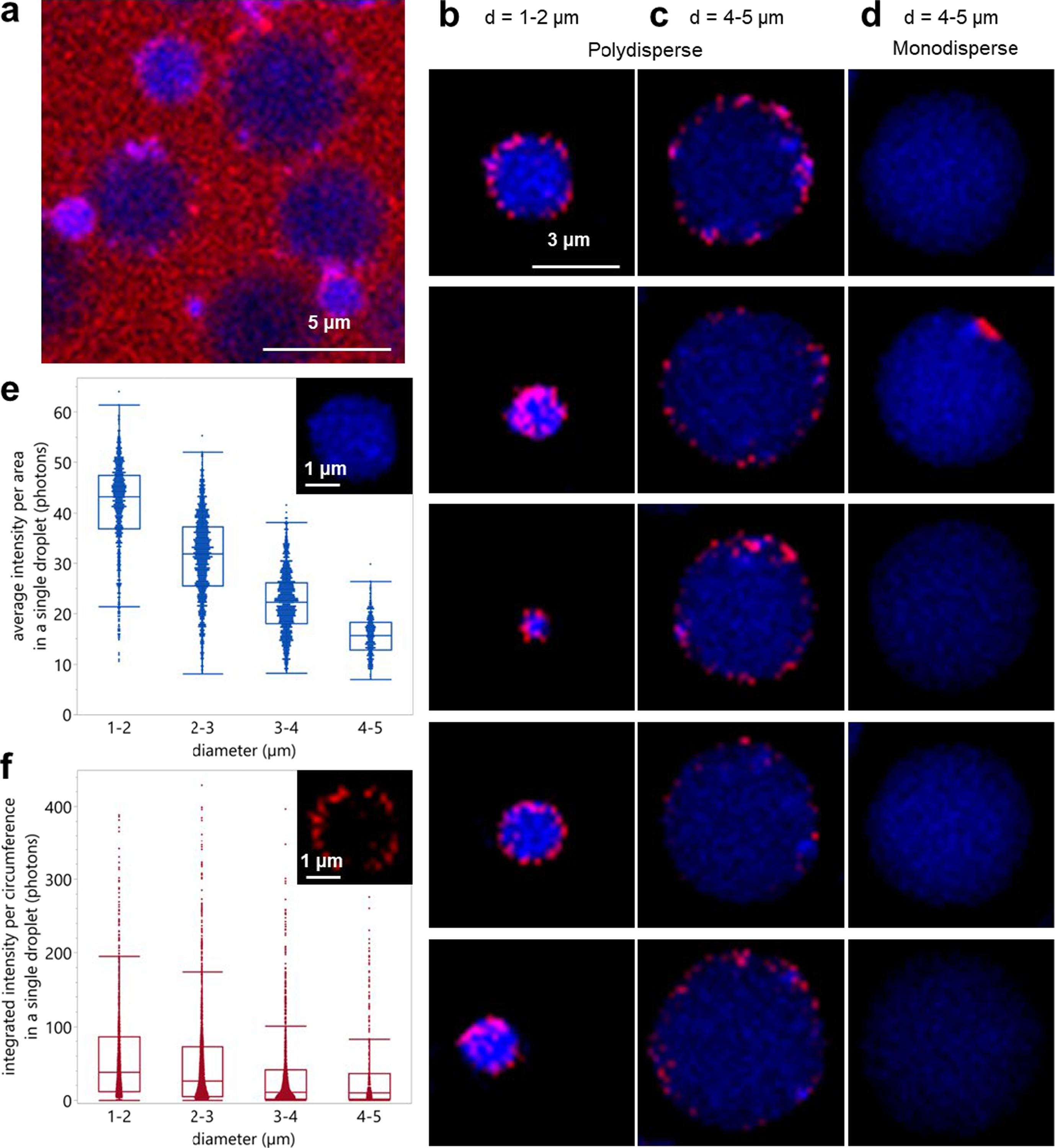
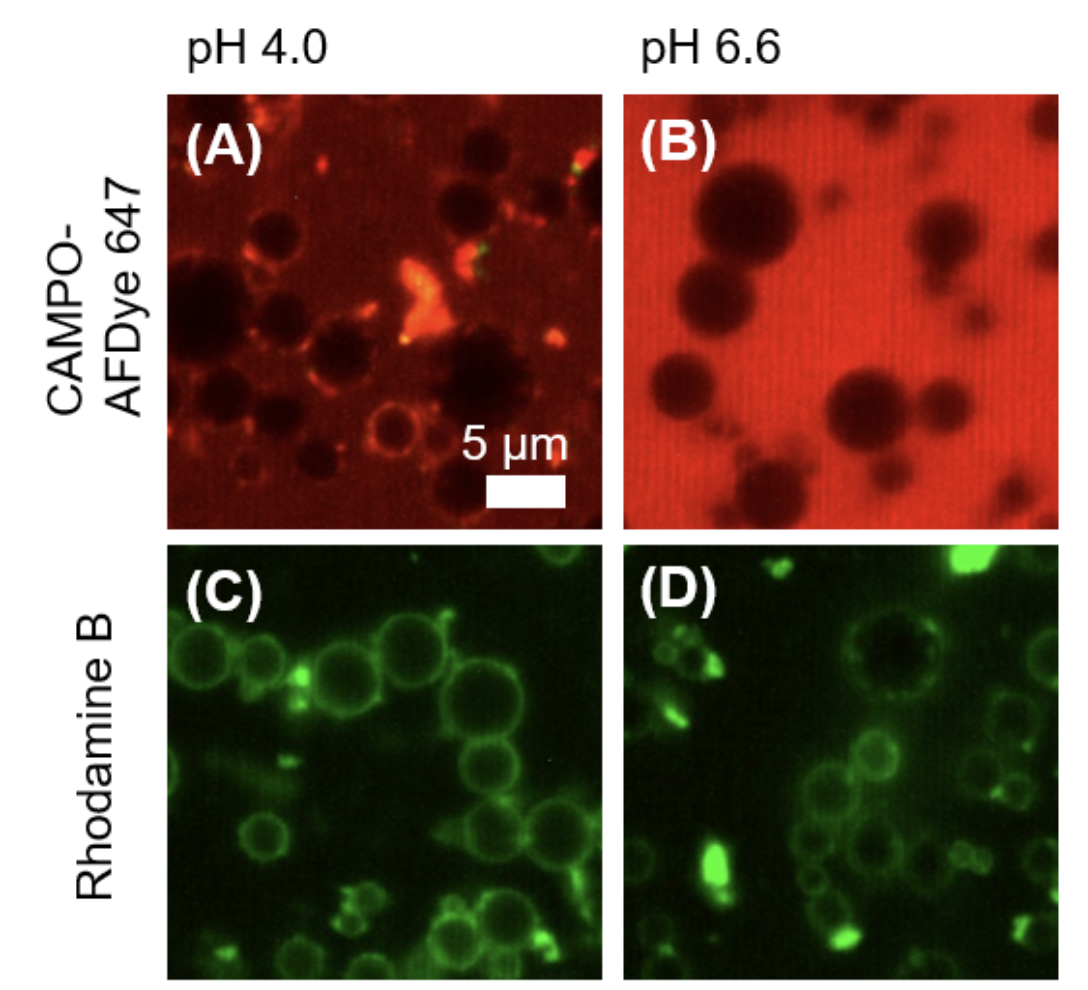
Spatiotemporal assessment of lipid and protein oxidation is key for understanding quality deterioration in emulsified food products containing polyunsaturated fatty acids. In this work, we first mechanistically validated the use of the lipid oxidation-sensitive fluorophore BODIPY 665/676 as a semi-quantitative marker for local peroxyl radical formation. Next, we assessed the impact of microfluidic and colloid mill emulsification on local protein and lipid oxidation kinetics in whey protein isolate (WPI)-stabilized emulsions. For that purpose, we also used BODIPY 581/591 C11 and CAMPO-AFDye 647 as colocalisation markers for lipid and protein oxidation. The polydisperse emulsions showed an inverse relation between droplet size and lipid oxidation rate. Further, we observed less protein and lipid oxidation occurring in similar sized droplets in monodisperse emulsions. This observation was linked to more heterogeneous protein packing at the droplet surface during colloid mill emulsification, resulting in larger inter-droplet heterogeneity in both protein and lipid oxidation. Our findings indicate the critical roles of emulsification methods and droplet sizes in understanding and managing lipid oxidation.